

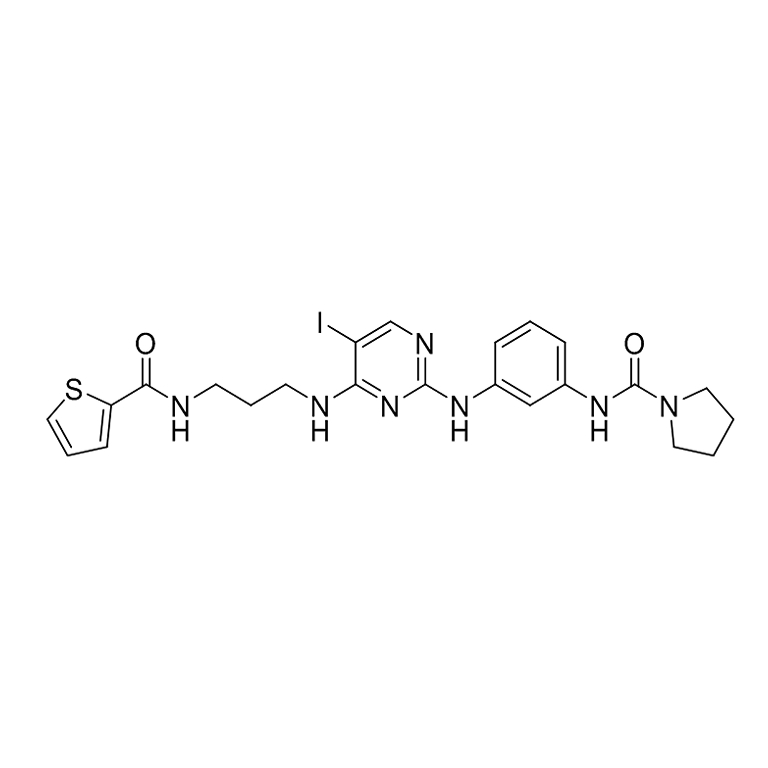
BX795
分子式:C23H26IN7O2S 分子量:591.47
|
产品描述 |
BX795是有效的,特异性PDK1抑制剂,IC50为6 nM,作用于PDK1比作用于PKA和PKC选择性分别高140和1600倍。 |
|||||
|
靶点 |
TBK1 |
IKKε |
PDK1 |
Aurora B |
ERK8 |
|
|
IC50 |
6 nM |
41 nM |
111 nM |
31 nM |
140 nM |
|
|
体外研究 |
BX795也抑制MARK1,MARK2,MARK4,NUAK1,VEGFR,MLK1,MLK2,和MLK3,IC50分别为55,53,19,5,157,50,46和42 nM。1 μM BX795不抑制如下酪氨酸蛋白激酶:肝配蛋白受体A2和B3,Syk,Bruton’s酪氨酸激酶,和FGFR1。然而,BX795抑制VEGFR,抑制效果比TBK1低。BX795抑制TBK1-催化的IRF3在Ser396位点磷酸化随着ATP浓度上升而下降,说明BX795是ATP竞争性抑制剂。BX795阻断TBK1和IKKε调节的IRF3激活,且抑制IFN-β的产量。聚(I:C)处理后,IRF3在核中积累,但是可被BX795抑制。BX795抑制IRF3依赖的基因转录。BX795抑制IFN-β从巨噬细胞中分泌。BX795对LPS刺激的p70核糖体S6激酶1在Thr229位点磷酸化没有效果, Thr229位点是PDK1作用靶点。BX795不影响IKKα/β复合体或LPS,聚(I:C),IL-1α,或TNFα促进的NFκB依赖的基因转录的激活。BX795作用于IL-1α或TNFα刺激的MEFs,也阻断JNK1/2和p38α MAPK磷酸化。BX795抑制TBK1/IKKε不会导致JNK1/2和p38α MAPK的激活。 |
|||||
|
体内研究 |
||||||
|
溶解性 |
DMSO:40mg/ml |
|||||
|
稳定性 |
2年-20°C粉状,6月-80°C溶于DMSO |
|||||
|
特征 |
||||||